Identifies genes that are significantly mutated in cancer genomes, using a model with mutational covariates.
Author: Mike Lawrence, et al., Broad Institute
Contact:
http://www.broadinstitute.org/cancer/cga/mutsig, http://www.broadinstitute.org/cancer/cga/cga_forums, mutsig-help@broadinstitute.org, http://genepattern.org/help
Algorithm Version: MutSigCV 1.3.01
Introduction
MutSigCV accepts whole genome or whole exome sequencing data from multiple samples, with information about point mutations, small insertions/deletions, and coverage, and identifies genes that are mutated more often than one would expect by chance.
Recent cancer genome studies have led to the identification of many cancer genes, and the expectation was that as samples sizes grew, the power to detect cancer driver genes (sensitivity) and distinguish them from the background of random mutations (specificity) would increase as well. However, it appears that one difficulty with larger sample size is that it increases the rate of detecting highly mutable genes as not simply highly mutable, but also implausibly cancer-related -- that is, it increases the false positive rate. One reason for this is the use of an average overall mutation rate for a given cancer type for the whole genome. In a mock case where the genes were given variable mutation frequencies and the dataset was analyzed under the erroneous assumption of a constant genome-wide mutation rate, many of the highly mutable genes were falsely detected as significant. The problem increases with sample size because the threshold for statistical significance decreases with increased sample size.
There are strong correlations between somatic mutation frequencies in cancers and both gene expression level and replication time of a DNA region during the cell cycle. Low-expressed and late-replicating genes, such as olfactory receptor genes and very large genes, make up many of the false positives seen in studies that are trying to identify cancer driver genes.
MutSigCV corrects for variation by employing patient-specific mutation frequencies and mutation spectra, and gene-specific mutation rates, incorporating expression levels and replication times. Incorporating these covariate factors into the model substantially reduces the number of false positives in the generated list of significant genes, especially when applied to tumor samples that have high mutation rates.
Algorithm
A critical component of MutSigCV is the background model for mutations, the probability that a base is mutated by chance. This model is not constant, but varies due to patient-based factors and genomic position-based factors. The patient-based factors include:
- overall mutation rate
- overall mutational spectrum (e.g., the percentages of mutations that are transitions of certain types, transversions of certain types, and/or nonsense)
The genomic position-based factors include:
- gene expression levels
- DNA replication timing
- HiC-based chromatin state estimation (that is, the degree to which the DNA is more or less densely packed, modulating transcriptional access to the genes in a given location)
The mutational spectrum model does not consider every possible base change on its own, but pools each mutation into categories that consider both sequence context (e.g., was this a mutation of a C that was adjacent to a G?) and functional impact (e.g., did this mutation create a stop codon? was it an indel?). Indels are counted as Null mutations.
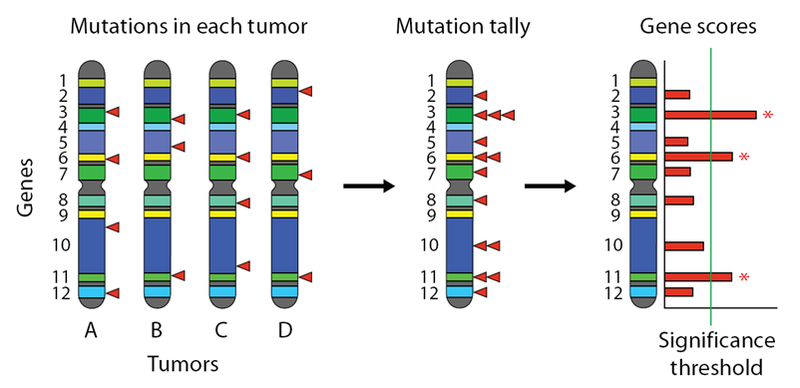
The following figure shows on the left a set of chromosomes, each from the tumor of a different cancer patient. Genes are cartooned as colored bands, and somatic mutations are indicated by red triangles. The mutations from all the tumors can be aggregated together by collapsing as shown, and the total number of mutations per gene can be computed. Then this tally is converted to a score, and then to a significance level. A threshold is chosen to control for the False Discovery Rate (FDR), and genes exceeding this threshold are reported as significantly mutated.
Usage
The MutSigCV module requires three files:
- MAF mutation file: A Mutation Annotation Format (MAF) file is a tab-delimited text file that lists mutations. The MAF file to be used in MutSigCV requires 2 additional, nonstandard columns, detailed in the Input Files section. If your MAF does not include these additonal columns it is possible for MutSig to automatically discover this information; for details, see below. This file could come from an analysis program like MuTect followed by Oncotater.
- Coverage file: A tab-delimited file that gives the maximum number of bases covered to adequate depth in order to call mutations. The coverage is broken down by gene and patient and also by mutation category. This file derives from processing sample coverage files in WIG (wiggle) format output by an analysis program like MuTect. If this level of detailed coverage information is not available, a file is available that will allow MutSigCV to operate assuming full coverage. See the Input Files section for details.
- Covariates file: The covariates table provided in the module (which can also be downloaded from the link in the Example Data section) was derived from multiple data sets. It has proven useful for analyzing many cancer types, but users can use more specific data if available.
For information on the contents and required formats of the input files see the Input Files section.
Note that though MutSigCV was developed for the analysis of somatic mutations, it has also been used successfully with germline mutations.
It has been observed that MutSigCV may not produce useful results on cancers with low mutation rates (such as pediatric cancers) due to certain internal assumptions made in the code. While a future version of MutSigCV may add the ability to change these assumptions before running the analysis, at present the GenePattern module is limited to using these defaults. It is possible to work around these assumptions, though you will need to obtain and modify the MatLab code and run it outside of GenePattern. Please contact the MutSigCV authors for more details.
References
Lawrence MS, Stojanov P, Polak P, Kryukov GV, et al. Mutational heterogenieity in cancer and the search for new cancer genes. Nature. 2013 Jun 16. doi: 10.1038/nature12213. (link)
The Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489:519-525. (link)
Parameters
| Name | Description |
|---|---|
| mutation table file * | Mutation list in Mutation Annotation Format (MAF). For more information on the file format, see the Input Files section. |
| coverage table file * | Coverage file in tab-delimited format, containing the number of sequenced bases in each patient, per gene per mutation category. For more information on the file format, see the Input Files section. |
| covariates table file * | Covariates table in a tab-delimited text file. For more information on the file contents and format, see the Input Files section. |
| output filename base * | Base name for the output files. |
| mutation type dictionary | The mutation type dictionary to use for automatic category and effect discovery. This is necessary only if you are using a MAF file without the columns "categ" and "effect". See the Input Files section for details. |
| genome build | Genome build to use for automatic category and effect discovery. This is necessary only if you are using a MAF file without the columns "categ" and "effect". See the Input Files section for details. |
* - required
Input Files
Note: gene and sample names must agree across these three files. Similarly, the categ numbers must agree between the mutation and coverage files.
- MAF file
The Mutation Annotation Format (MAF) file is a tab-delimited text file that contains information about the mutations detected in the sequencing project. It lists one mutation per row, and the columns (named in the header row) report several pieces of information for each mutation. One common source for MAF files that have been used in MutSigCV during the algorithm's development was the MuTect tool, followed by annotation of its output using Oncotator. More information can be found on MuTect here: http://www.broadinstitute.org/cancer/cga/mutect. Information about Oncotator can be found here: http://www.broadinstitute.org/oncotator.
The MAF file to be used in MutSigCV must have 2 additional, nonstandard columns: effect and categ. MutSigCV requires only 4 columns of the MAF file (see this page for the full MAF specification) and can process a simple tab-delimited file with only these columns if a full MAF is not available. The columns are:
- gene: Name of the gene that the mutation was in (can also be called "Hugo Symbol")
- patient: Identifier for the sample with the mutation (can also be called "Tumor Sample Barcode").
- effect: Indicates whether the mutation is located within an exon (or splice site), or outside the coding regions, and whether the mutation is silent or not if it is located in a coding region; this information can be derived from the standard Variant_Classification MAF column.
- categ: The mutation category. Encodes both base context and what type the mutation was. This column can be derived from three of the columns from a standard MAF file, Variant_Classification, Reference_Allele, and Tumor_Seq_Allele1, along with other genomic information to identify CpG contexts and null mutations. Each mutation belongs to only one category. If it is a null mutation or indel, it belongs in the last category.
- CpG transitions
- CpG transversions
- C:G transitions
- C:G transversions
- A:T transitions
- A:T transversions
- null+indel mutations
Note that if your MAF does not contain category and effect information, you can direct MutSigCV to use its preprocessor to automatically organize these columns. To do so, you need to provide a mutation type dictionary and tell it which genome build to use. A suitable mutation type dictionary can be found on the public GenePattern server using "Add Path or URL" under "shared_data/example_files/MutSigCV_1.3/mutation_type_dictionary_file.txt". Category and effect discovery is only available for human genomes hg19 and hg18 at present.
- Coverage file
This file contains information about the sequencing coverage achieved for each gene and patient/tumor. Within each gene-patient bin, the coverage is broken down further according to the mutation category (e.g., A:T basepairs, C:G basepairs), and also according to the effect (silent/nonsilent/noncoding). This tab-delimited file can be produced by processing the sample-level coverage files in WIG (wiggle) format output by the MuTect tool. More information on MuTect can be found here: http://www.broadinstitute.org/cancer/cga/mutect. If detailed coverage information is not available, the user can use a “full coverage” file that is available on the GenePattern server.
The columns of the file are:
- gene: name of the gene for which this line reports coverage (corresponds to the MAF file's Hugo_Symbol)
- effect: silent, nonsilent, or noncoding
- categ: number of the category that this line reports coverage for (must match the categ in the mutation table)
- <patient_name_1>: number of covered bases for this gene, effect, and category
- <patient_name_2>: number of covered bases for this gene, effect, and category
- <patient_name_ ...>: number of covered bases for this gene, effect, and category
Note, covered bases will typically contribute fractionally to more than one effect depending on the consequences of mutating to each of three different possible alternate bases.
We recognize that detailed coverage information is not always available. In such cases, a reasonable approach is to carry out the computation assuming full coverage. The MutSigCV developers have prepared a file that can be used for this purpose: it is a "full coverage" file, or more accurately a "territory" file: the only information it contributes is a tabulation of how the reference sequence of the human exome breaks down by gene, categ, and effect. On the GenePattern public server, this file can be found using "Add Path or URL" under "shared_data/example_files/MutSigCV_1.3/exome_full192.coverage.txt".
- Covariate file
This file contains the genomic covariate data for each gene, for example, expression levels and DNA replication times, that will be used in MutSigCV to judge which genes are close to each other in mathematical "covariate space."
In general, the columns of this file are:
- gene: name of the gene for which this line reports coverage
- COVARIATE1 NAME: value of COVARIATE1 for this gene
- COVARIATE2 NAME: value of COVARIATE2 for this gene
- COVARIATEnv NAME: value of COVARIATEnv for this gene
For the specific data file supplied in GenePattern, the columns are:
- gene: name of the gene for which this line reports coverage
- expr: expression level of this gene, averaged across 91 cell lines in the Cancer Cell Line Encylcopedia (CCLE)
- reptime: DNA replication time of this gene (measured in HeLa cells), ranging from 100 (very early) to 1000 (very late)
- hic: chromatin state of this gene (measured from HiC experments in K562 cells) ranging approximately from -50 (very closed) to +50 (very open)
Output Files
- <output.filename.base>.sig_genes.txt MutSig report
A tab-delimited report of significant mutations, listed in descending order from most significant to least significant.The "nnei","x", and "X" values in the MutSig output analysis give insight into how the background mutation rate is calculated for a given gene. nnei gives the number of neighboring genes that are pooled together to compute the background mutation rate for that gene; these genes are not necessarily adjacent on the genome, but rather they have nearby covariate values. x gives the number of mutated bases in these neighboring genes that are either silent or non-coding, while X gives the total number of bases related to these neighboring genes.
Example Data
The data used for the TCGA Lung Squamous paper is available here:
LUSC.MutSigCV.input.data.v1.0.zip
Requirements
MutSigCV can only be used on the GenePattern public server, as it requires a specialized installation process that prevents distribution via the repository. Please contact the authors listed above if you have an interest in installing MutSigCV locally.
Acceptance of the module license is required for its use. A copy of the license text is available here: www.broadinstitute.org/cancer/cga/sites/default/files/data/tools/mutsig/mutsig_public_license.html
Platform Dependencies
Task Type:
MutSig
CPU Type:
any
Operating System:
Linux
Language:
MATLAB 2013a
Version Comments
| Version | Release Date | Description |
|---|---|---|
| 1 | 2013-06-17 |